


▲共同第一作者:刘文博,李健斌;通讯作者:李朝军通讯单位:加拿大麦吉尔大学论文DOI:10.1002/anie.201909138由于芳环的普遍性,亲核试剂与芳香族亲电试剂(Csp2-X)交叉偶联反应在有机化学中占有重要地位。与脂肪族亲电试剂(Csp3-X)不同,芳基亲电试剂无法通过 SN1 或 SN2 历程与亲核试剂(Nu)进行反应。这是因为芳基亲电试剂的 σ*c-x 反键轨道不容易接近, 很难与亲核试剂进行有效的作用(图1A)。在现代方法学中,过渡金属催化剂的出现极大地突破了亲核试剂与芳香亲电试剂交叉偶联反应的限制。通过过渡金属的氧化加成来活化芳基亲电试剂的 Csp2-X 键,经过转金属化和还原消除后,芳基亲电试剂和亲核试剂的交叉偶联得以实现。基于这种反应机理,生成 C-C,C-O,C-N,C-S,C-P,C-B 和其他键的交叉偶联反应已被实现(图1B)。尽管过渡金属能够有效地催化这些偶联反应,它们也带来了其他的问题。例如,贵金属,钌和铑比较昂贵且带有毒性。在大规模的合成应用中,过渡金属催化剂价格和产物纯化往往加重了成本负担。而且,过渡金属通常需要配体的协助,通过形成金属配合物来获得良好的催化效果。在许多情况下,配体甚至比金属本身的价格还要高。此外,由于一些金属配合物对水和空气敏感,这导致操作上的不便以及实验安全上的考虑。为了解决这些问题,一种比较理想的方法便是发展无需金属催化剂参与的亲核试剂与芳香亲电试剂交叉偶联反应。在这个背景之下,光化学被认为是一种具有广阔前景的工具(图1C)。在基态下,尽管部分芳基亲电试剂(例如具有吸电子基团的底物)可以通过SNAr反应与亲核试剂进行反应,芳基亲电试剂通过直接加成/消除机理通过形成非芳香性的Meisenheimer中间体与亲核试剂反应需要跨过相当高的能垒。而在涉及过渡金属催化剂的路径中,过渡金属则可以通过形成金属-底物加合物来改变反应历程从而降低反应的能垒。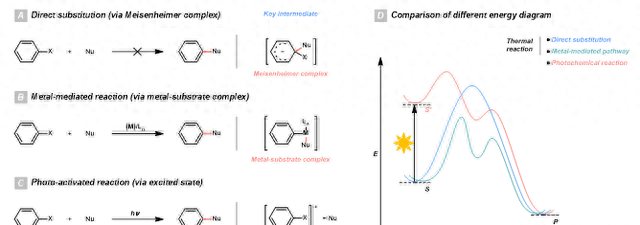 ▲图1
▲图1
与过渡金属化学有所不同,光化学利用光来激发底物。通过吸收光子,分子会被激发到更高的能态。这为随后的众多反应模式奠定了基础,并为实现一些有趣的化学转化提供了机会。而这种反应活性是当底物在基态时往往是难以实现的。在吸收光子后,处于激发态的分子显示出两个明显的特征:一方面,被激发的芳基亲电试剂中,Csp2-X 键能被削弱,容易导致 C-X 的均裂或者异裂;另一方面,与基态的分子相比,光子受体的氧化还原性质发生了变化。受激分子有两个半满的分子轨道。
与基态相比,最高占据分子轨道(HOMO)能量变得更高,而最低未占据分子轨道(LUMO)能量有所下降,从而使其同时具有更强的氧化性和还原性。在过去的十年中,通过使用外加的光敏剂进行光催化氧化还原反应已经引起了广泛的关注。与这种需要外加光敏剂的光催化反应相辅相成的另一类光反应则是通过光直接激发底物从而诱导所设计的反应发生。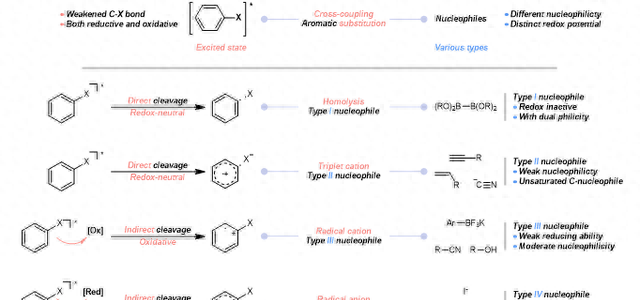 ▲图2
▲图2
在无金属参与的芳基取代和交叉偶联反应中,可以设想,由于具有了上述的电子构型变化,芳基亲电子试剂在激发态下会表现出新颖独特的反应活性。从机理上分析,按照具体的反应条件以及亲核底物的性质,激发后的芳基亲电试剂可以通过以下四种模式来进行反应(图2):(A)激发后,被激发的芳基亲电试剂可能会发生均裂并直接生成芳基自由基进行反应;(B)通过离去(拟)卤素阴离子,激发态下的亲电试剂可以产生高活性的芳基阳离子从而与一系列惰性亲核试剂偶联;(C)由于在激发态下同时具有氧化还原两种能力,通过单电子转移(SET),芳基亲电试剂可以被氧化成芳基自由基阳离子;(D)通过接受电子,还原为自由基阴离子并与其他亲核反应物进行结合。与之相对应,亲核试剂根据其固有的亲核性质和氧化还原电位可也以分为四种类型。该综述近期发表在Angewandte Chemie International Edition上。文章的共同第一作者是博后刘文博和博士研究生李健斌,第二作者为博士研究生Chia-Yu Huang。该论文:Liu, W.; Li, J.; Huang, C. Y.; Li, C. J., Aromatic Chemistry in the Excited State: Facilitating Metal-Free Substitutions and Cross-Couplings. Angew. Chem., Int. Ed. 2019, DOI: 10.1002/anie.201909138课题组介绍详见李朝军课题组主页:http://www.cjlimcgill.ca/研之成理各版块内容汇总:1.
仪器表征基础知识汇总
2. SCI论文
写作专题汇总
3.
Origin/3D绘图等科学可视化汇总
4.
理论
化学
基础知识汇总
5. 催化板块汇总
6. 电化 – 电池相关内容汇总贴
7. 研之成理名师志汇总
更多科研作图、软件使用、表征分析、SCI 写作、名师介绍等干货知识请进入研之成理后台自主查询。
本网页内容旨在传播知识,若有侵权等问题请及时与本网联系,我们将在第一时间删除处理。E-MAIL:dandanxi6@qq.com